News
Events
Directory
About
At a Glance
Get Involved
Institutes & Centers
History
Employment
Supporting Our Community
People
Everyone
Faculty
Administrative Staff
Academics
Undergraduate
Graduate
Resources
Departmental Services
Faculty Affairs
Research & Facilities
Room Reservations & Events
Resources Calendar
Seminar Archive
IB Wiki
BIO Undergrad Info
IB Administrative Portal
Contact
Contact
JavaScript is currently disabled.
Please enable it for a better experience of
Jumi
.
The
Biodiversity Center
is one of many affiliated centers and institutes.
From Genes to Ecosystems
, we study organisms from their molecular base to their global interconnections
Faculty Leaders
include some of the most cited researchers in the world. Four are members of the National Academy of Sciences
Graduate education
programs ranked among the top 10 nationally
Research strengths
include animal behavior, bioinformatics, genomics, infectious disease, microbiomes and more
Featured
More News »
Study Challenges Popular Concept of Spread of Cultural Innovations
Red Flags: I’m Not the Bug for You!
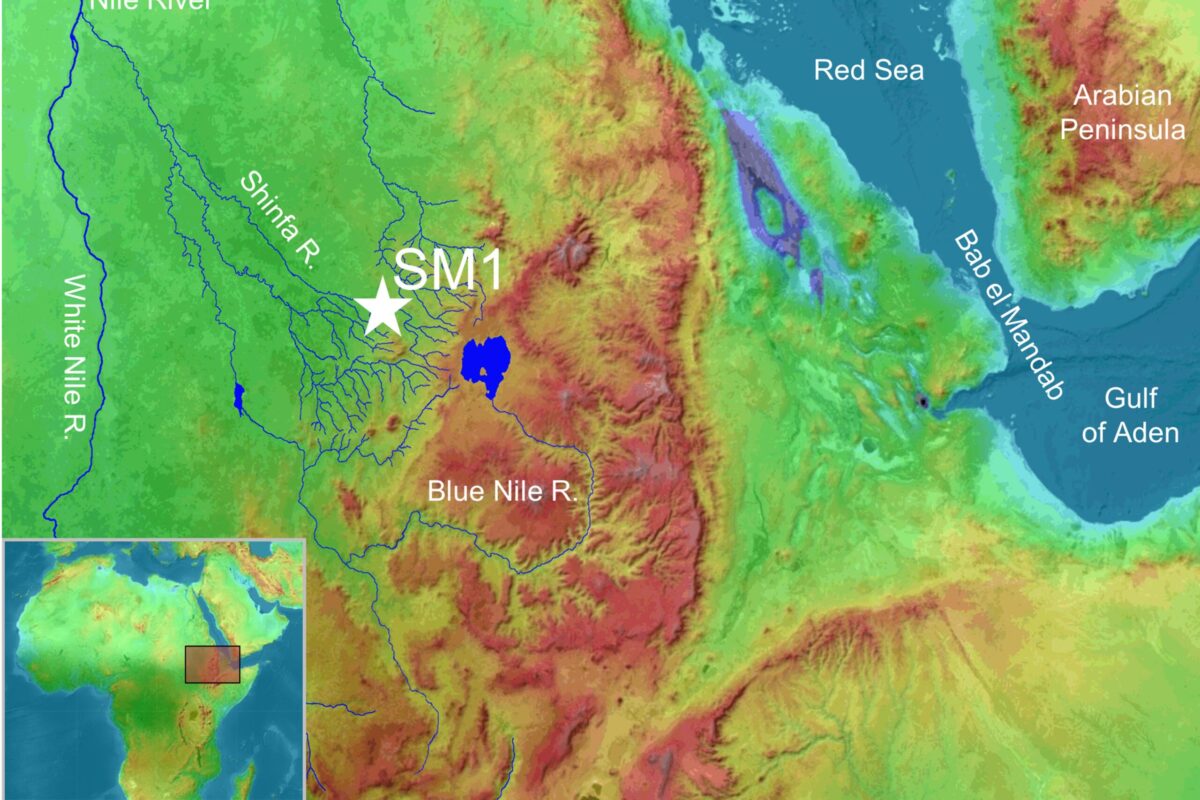
Surviving a Volcanic Supereruption May Have Facilitated Human Dispersal Out of Africa
A Once-in-Many-Centuries Event
Events
29
Apr
Mon Apr 29 @12:00AM
Last class day
29
Apr
Mon Apr 29 @ 3:00PM
-
04:00PM
IB Seminar: Edmund Basham, Stengl-Wyer Scholar
01
May
Wed May 01 @ 8:00AM
-
05:00PM
Integrative Biology Retreat (All Day)
View Full Calendar
EXPLORE
Faculty »
Employment »
Resources »
Biodiversity Center »
Visitors »
Seminars »
Field Stations »
Institutes & Centers »
JavaScript is currently disabled.
Please enable it for a better experience of
Jumi
.